DIAGNÓSTICO: Distrofia corneal granular
Descripción
macroscópica: Se recibe un botón corneal de 9 mm de diámetro con
opacidad amarillenta central, conservando la transparencia a nivel
periférico.
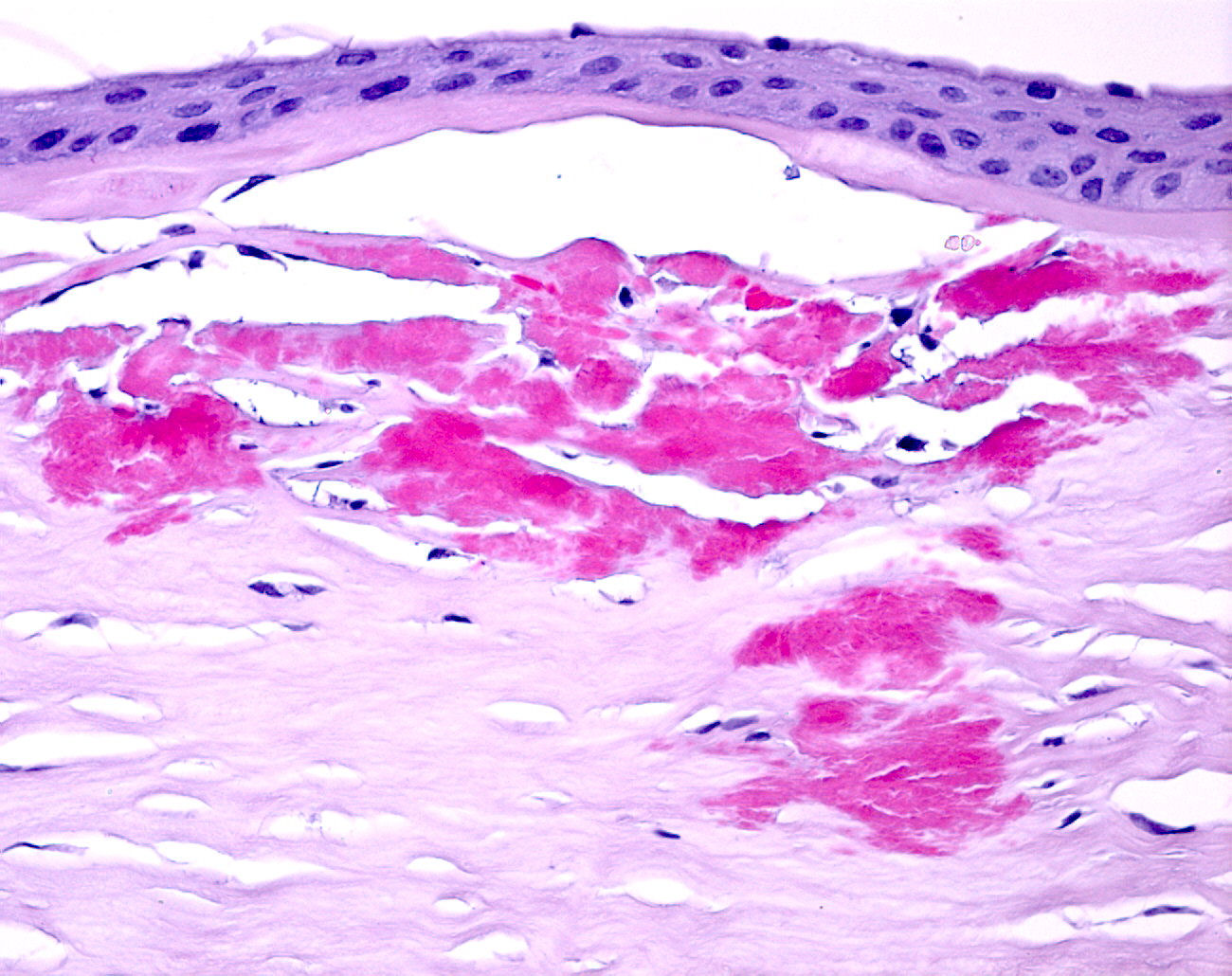
Descripción microscópica y diagnóstico: Se observa una cornea
de epitelio edematoso y de grosor variable, con abundantes bullas
subepiteliales. La capa de Bowman se halla ausente de manera focal.
A nivel del estroma superficial se observan depósitos de un material
eosinófilo granular, negativo para las técnicas histoquímicas de
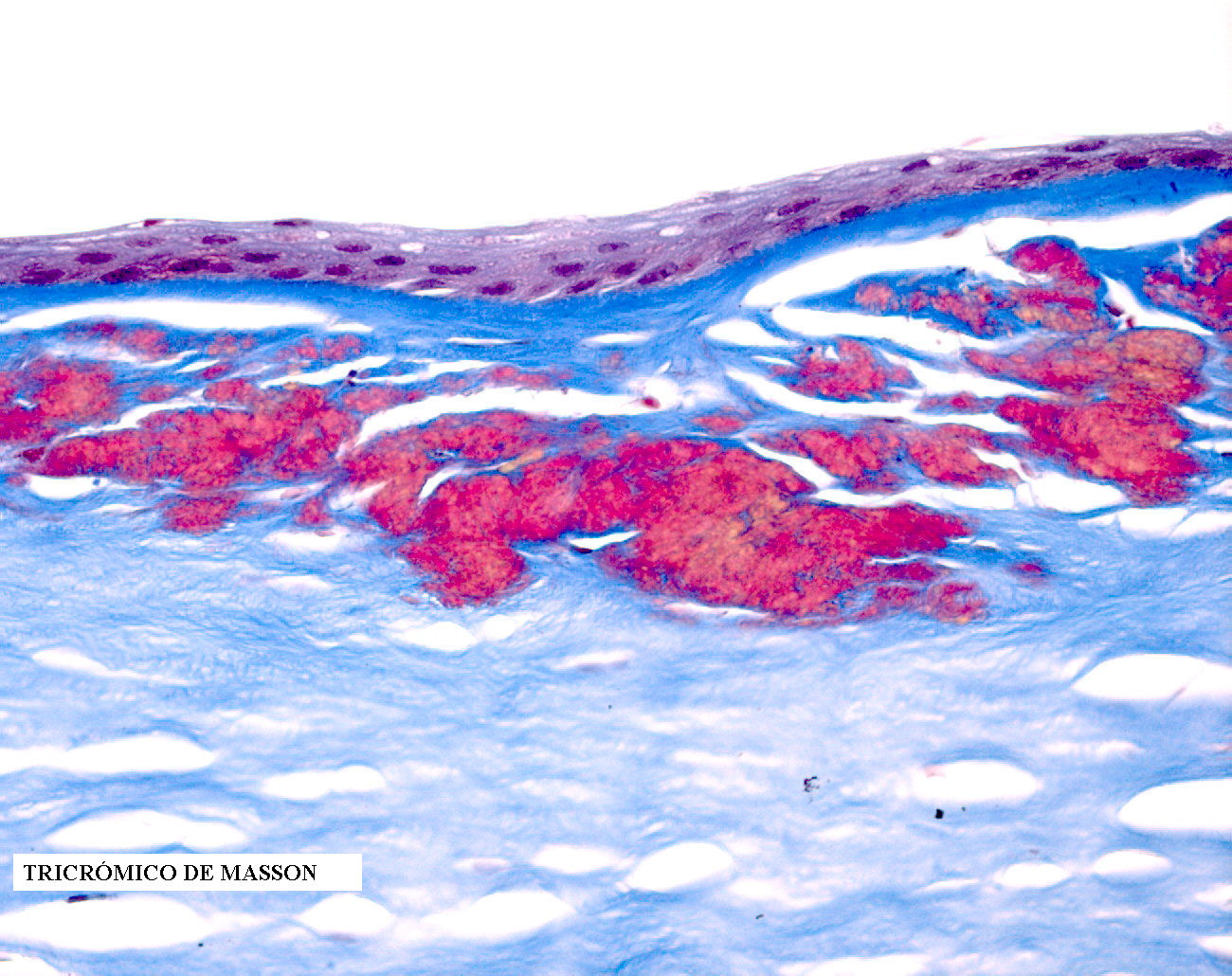
azul alcián y rojo congo, siendo en cambio positivas para el
tricrómico de Masson que tiñe en rojo los depósitos. No se observa
estroma posterior, membrana de Descemet ni endotelio, tal como
corresponde a una queratectomía superficial.
Los hallazgos
histológicos son compatibles con una distrofia corneal estromal
de tipo granular.
Discusión: En la actualidad las distrofias son consideradas
defectos localizados hereditarios del nacimiento que no se ponen de
manifiesto clínicamente hasta la madurez. Característicamente son
lesiones bilaterales y simétricas(1).
La distrofia
granular es una lesión hereditaria, con patrón autosómico dominante
localizada en el cromosoma 5(2) por mutación del gen BIGH3(3). Suele
aparecer desde la niñez o adolescencia como opacidades granulares
focales blanco-grisáceas localizadas en el estroma superficial. La
periferia de la córnea suele permanecer clara, lo mismo que el
estroma entre las opacidades granulares. A medida que la enfermedad
avanza las opacidades aumentan en número y tamaño, y comienzan a
cohalescer o hacerse más profundas, aunque el estroma profundo
raramente se afecta. El curso es lento y, debido a la no afectación
corneal entre las opacidades, la pérdida visual significativa no
sucede hasta la cuarta o quinta época(4). Ocasionalmente las
opacidades se proyectan hacia el epitelio y pueden causar erosiones
con episodios recurrentes de irritación, fotofobia y congestión
perilímbica. Esta distrofia recurre frecuentemente en el injerto, en
caso de tratamiento con injerto corneal, adoptando la misma
morfología que la enfermedad inicial(5).
Excepciones al
curso clínico descrito incluyen una forma de distrofia granular
superficial confluente que se sitúa en el área central dejando un
anillo periférico no afecto, como en este caso, así como una forma
rápidamente progresiva en pacientes sin historia familiar. En estos
casos los depósitos se suelen localizar cerca de la capa de Bowman,
causando confusión inicial con la distrofia de Reis-Büclers.
La microscopía
óptica típica consiste en depósitos de un material eosinofílico en
el estroma, que no son birrefringentes y tiñen en rojo brillante con
la técnica del tricrómico de Masson, pudiendo teñir menos
intensamente con el PAS de lo que lo hace el estroma normal. Estos
depósitos son negativos para mucopolisacáridos ácidos (azul alcián)
o amiloide (rojo congo), lo que distingue a esta entidad de otras
distrofias estromales como la distrofia macular o la distrofia de
Lattice respectivamente.
Histoquímicamente los depósitos parecen ser una proteína no colágena
que contiene tirosina, arginina y aminoácidos sulfuros y, aunque la
fuente de este material es desconocida, algunos autores sugieren un
origen en el epitelio corneal (6).
Bibliografía:
1. Ophthalmic Pathology, an atlas and textbook. William H.
Spencer, MD. 4th edition. 1997. Ed. Saunders.
2. Klintworth GK. The molecular genetics of the corneal dystrophies--current
status.Front Biosci. 2003 May 1;8:687-713.
3. Li Y, Sun XG, Ren HY, Dong B, Wang ZQ, Sun XY. Analysis of human
transforming growth factor beta-induced gene mutation in corneal
dystrophy. Chin Med J (Engl). 2004 Sep;117(9):1418-21.
4. Collin HB, Hendicott PL. Granular dystrophy of the cornea. Clin
Exp Optom. 1999 Sep;82(5):203-206.
5. Marcon AS, Cohen EJ, Rapuano CJ, Laibson PR. Recurrence of
corneal stromal dystrophies after penetrating keratoplasty. Cornea.
2003 Jan;22(1):19-21.
6. Wollensak G, Witschel H. Vimentin and cytokeratin pattern in
granular corneal dystrophy. Graefes Arch Clin Exp Ophthalmol. 1996
Aug;234 Suppl 1:S110-4.